
How FUS Fits into the Bigger RNA Protein Picture
January 3, 2023
While the precise mechanisms that lead to the development of amyotrophic lateral sclerosis (ALS) are still under investigation, several genetic mutations have been linked to the disease and its symptoms. By investigating these mutations, researchers hope to discover therapeutic interventions that can slow down or stop the neurodegenerative effects of ALS.
Recently, researchers have found possible connections between hundreds of genes and ALS. Many of which may provide targets for new treatments.1 But there are a handful of well-researched genes that researchers believe mutate in a way that causes their proteins to fold incorrectly in the body’s neurons.2 The investigation of these genes and the development of ribonucleic acid (RNA) therapies for ALS and other neurodegenerative diseases is one of the most exciting developments in the field.

How FUS Fits into the Bigger RNA Protein Picture
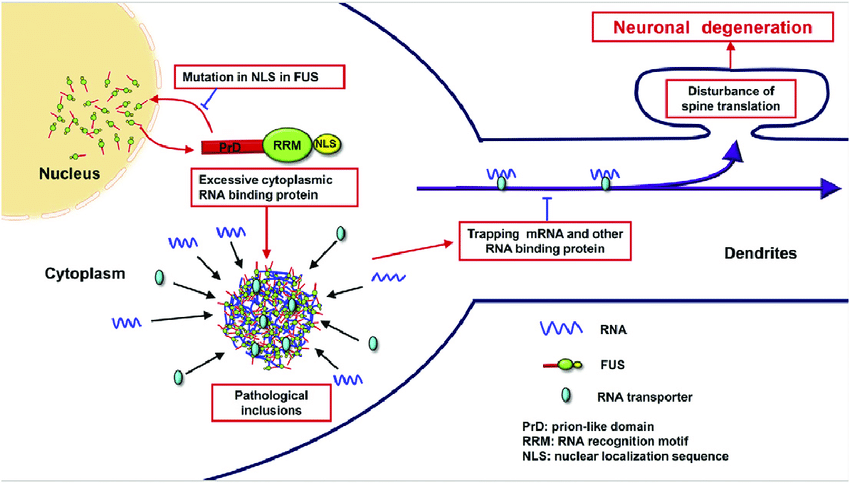
A handful of gene mutations are associated with ALS. Among them is the gene that produces the RNA-binding protein called fused in sarcoma (FUS). A mutation in the FUS gene can lead to familial form of ALS, while FUS mislocalization and proteinopathy can be involved in both sporadic and familial cases of ALS.
The FUS protein is associated with a rare but aggressive form of ALS (FUS-ALS) as well as frontotemporal dementia. When FUS binds to DNA in specific genes that are transcribed by RNA polymerases II and III, it can result in changes in transcription levels. FUS also controls protein production by regulating pre-mRNA processing and helping to transport mRNAs out of the nucleus into the cytoplasm.
Researchers continue to make progress in identifying the precise role of FUS in ALS in the hopes of creating gene therapies that can address symptoms of the disease.
What is the Purpose of FUS?
FUS has many physiological functions, most of which researchers are still trying to understand fully. What we do know is that FUS regulates multiple aspects of RNA metabolism and DNA damage repair.1
Note. Shiihashi, G. Et al. (2017) Dendritic Homeostasis Disruption in a Novel Frontotemporal Dementia Mouse Model Expressing Cytoplasmic Fused in Sarcoma. EBioMedicine. https://www.researchgate.net/publication/319617412_Dendritic_Homeostasis_Disruption_in_a_Novel_Frontotemporal_Dementia_Mouse_Model_Expressing_Cytoplasmic_Fused_in_Sarcoma
FUS is an exporter of mRNA from the nucleus to the cytoplasm. When FUS proteins are attached to pre-mRNA, this typically serves as a signal that the transcription process is not complete and the mRNA should not yet be exported from the nucleus to the cytoplasm. The protein also plays a crucial role in DNA repair. When DNA is damaged, it is one of the first proteins recruited to the site. Therefore, any impairment in the production of FUS can have a harmful effect on DNA repair.
The FUS protein consists of 15 exons that encode 526 amino acids. It is a critical component of the heterogeneous nuclear ribonucleoprotein (hnRNP) complex, which is present in a cell’s nucleus during gene transcription. Due to its abundance in the nucleus, FUS forms stable complexes with hnRNP components.2
FUS also helps regulate transcription by interacting with transcription factors and transcription initiation factor TFIID. It can recruit and bind to RNA polymerase II and then alter its phosphorylation status.3
Where is FUS Found?
The FUS gene is located on chromosome 16 (its specific location is 16p11.2). Interestingly, when this region is deleted, it can lead to problems with learning and language.
FUS protein is found in higher levels in neurons than other cell types. While Fus is typically found in both the cytoplasm and nucleus in neurons, it is found mainly in the nucleus in glial cells.4 High expression in neurons helps explain why FUS may play a role in neuronal plasticity and the maintenance of dendritic integrity.
Is FUS Similar to TDP-43?
FUS and TAR DNA binding protein 43 (TDP-43) are both DNA/RNA binding proteins with similar structures and functions. Misfolded FUS and TDP-43 proteins or abnormal cellular aggregation of their proteins are also hallmarks of neurodegenerative disorders such as ALS and frontotemporal dementia (FTLD). TDP-43 buildup occurs in about 97% of people with ALS, even patients without apparent genetic mutations.
Pathological accumulations of both TDP-43 and FUS have also been found in the cytosol of ALS patients.5 When this happens, it generally means there are fewer of these proteins in the nucleus to do their jobs. Therefore, failed nuclear import mechanisms are another avenue for exploration.
The ubiquity of TDP-43 and FUS proteinopathies in ALS patients makes both proteins potential targets for new gene therapies.6 Target ALS is helping to fund research into both. Related published research can be found here:
doi: 10.1016/j.celrep.2019.07.069
doi: 10.1038/ncomms10465
doi: 10.1172/JCI139741
What is the Role of FUS in ALS?
Since the FUS protein plays a role in cellular processes ranging from DNA repair to RNA splicing and transport, it can trigger a wide variety of toxic effects in the body if it’s deleted or accumulates in the wrong place.
Researchers have also hypothesized that FUS plays a role in neuronal plasticity and dendritic integrity.7
The overaccumulation of FUS proteins in the cytoplasm may lead to toxic FUS gene mutations linked to ALS. While the exact mechanisms are still being researched, experiments to help regulate FUS in the cytoplasm have shown some promise for decreasing the lethality of some ALS cases.8
Researchers have also demonstrated that non-allele-specific FUS antisense oligonucleotide (ASO) therapies FUS silencing can reduce the toxic accumulation of FUS protein in the brain and spinal cord and delay motor neuron degeneration in FUS-dependent ALS.9
The Future of FUS-ALS Research
Researchers continue to expand their knowledge of ALS genetics in the hopes of developing novel gene therapies for the disease. FUS mutations cause a particularly severe form of ALS, so while it may not play a role in a large number of cases, these gene therapies may put an end to much suffering.
Currently, Target ALS funds multiple projects on the role of FUS proteins in the disease as well as the role of novel therapies like viral vectors in decreasing mutations in the FUS gene or correcting the type of problematic FUS expression that is linked to neurodegenerative diseases.
Frequently Asked Questions
What percent of ALS cases involve FUS?
About 1% of total ALS cases are associated with FUS protein aggregation and about 5% of fALS cases are associated with a FUS mutation.10
Is FUS a transcription factor?
FUS is a multifunctional protein that plays a role in transcriptional regulation and mRNA processing. It binds to DNA to make new proteins and splices mRNA before transporting it out of the nucleus to the cytoplasm in the cells, where it can be further processed by other structures. FUS can also help in DNA repair to prevent genetic damage.
While FUS may aid in transcriptional activation and is associated with gene-specific transcription factors, FUS itself is not a transcription factor. Transcription factors are the specific proteins that help transcribe DNA into RNA.
Sources:
1. Chen, C. et al. (2019, 24 Apr). Fused in sarcoma: Properties, self-assembly, and correlation with neurodegenerative diseases. Molecules. https://pubmed.ncbi.nlm.nih.gov/31022909/
2. Zinszner, H. et al. (1997). TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. Journal of Cell Science. https://www.researchgate.net/profile/Helene-Zinszner/publication/13956708_TLS_FUS_binds_RNA_in_vivo_and_engages_in_nucleo-cytoplasmic_shuttling/links/02e7e532b04984a2a9000000/TLS-FUS-binds-RNA-in-vivo-and-engages-in-nucleo-cytoplasmic-shuttling.pdf
3. Chen, C. et. al.
4. Rademakers, R. et al. (2011, 1 Aug). FUS gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2969843
5. Dormann, D. et al. (2011). TDP-43 and FUS: a nuclear affair. Trends in Neurosciences. https://pubmed.ncbi.nlm.nih.gov/21700347/
6. Mackenzie, I. et al. (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia.” The Lancet. Neurology. https://pubmed.ncbi.nlm.nih.gov/20864052
7. Sanjuan-Ruiz, I., Govea-Perez, N., et al. (2021, Sep 6) Wild-type FUS corrects ALS-like disease induced by cytoplasmic mutant FUS through autoregulation. Molecular Neurodegeneration. https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/s13024-021-00477-w
8. Sanjuan-Ruiz, I., et al.
9. Korobeynikov, V. A. (2022, January 24). Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nature. https://www.nature.com/articles/s41591-021-01615-z
10. Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013 Aug 7;79(3):416-38. doi: 10.1016/j.neuron.2013.07.033. PMID: 23931993; PMCID: PMC4411085.
Related News

Dear Dad
May 21, 2026

All In for Margy
May 7, 2026


Q1 2026 Publications from our Innovation Ecosystem
March 31, 2026